Fenilcetonuria
La fenilcetonuria, conocida también por sus siglas PKU (del inglés Phenylketonuria), es una enfermedad genética hereditaria que afecta la forma en que el organismo procesa un aminoácido llamado fenilalanina. Este aminoácido se encuentra en muchos alimentos que contienen proteínas, como la carne, el pescado, los huevos, la leche, los quesos y algunas legumbres. Cuando una persona tiene fenilcetonuria, su cuerpo no produce suficiente cantidad de la enzima necesaria para descomponer la fenilalanina, lo que provoca que esta sustancia se acumule en la sangre y en los tejidos.
La enfermedad se transmite de padres a hijos mediante un patrón hereditario autosómico recesivo. Esto significa que para desarrollar la enfermedad, una persona debe recibir una copia alterada del gen de cada uno de sus padres. Los padres generalmente no presentan síntomas porque son portadores sanos de una sola copia del gen alterado.
La fenilalanina es un nutriente esencial para el organismo, pero cuando se acumula en niveles elevados puede resultar tóxica, especialmente para el cerebro en desarrollo. Si la enfermedad no se detecta y trata de manera temprana, puede causar daño neurológico irreversible. Por esta razón, la fenilcetonuria es una de las enfermedades incluidas en los programas de tamiz neonatal en muchos países.
Los recién nacidos con fenilcetonuria suelen parecer completamente sanos al nacer. Sin embargo, con el paso de los meses pueden comenzar a presentar retraso en el desarrollo, dificultades para aprender, irritabilidad, hiperactividad, problemas de comportamiento, convulsiones y discapacidad intelectual. También pueden presentar piel clara, cabello rubio y ojos claros debido a alteraciones en la producción de melanina.
Otros signos frecuentes incluyen un olor característico en la piel, la orina o el aliento, causado por la acumulación de ciertos compuestos derivados de la fenilalanina. Además, algunos pacientes pueden experimentar problemas de crecimiento y alteraciones neurológicas si no reciben tratamiento adecuado.
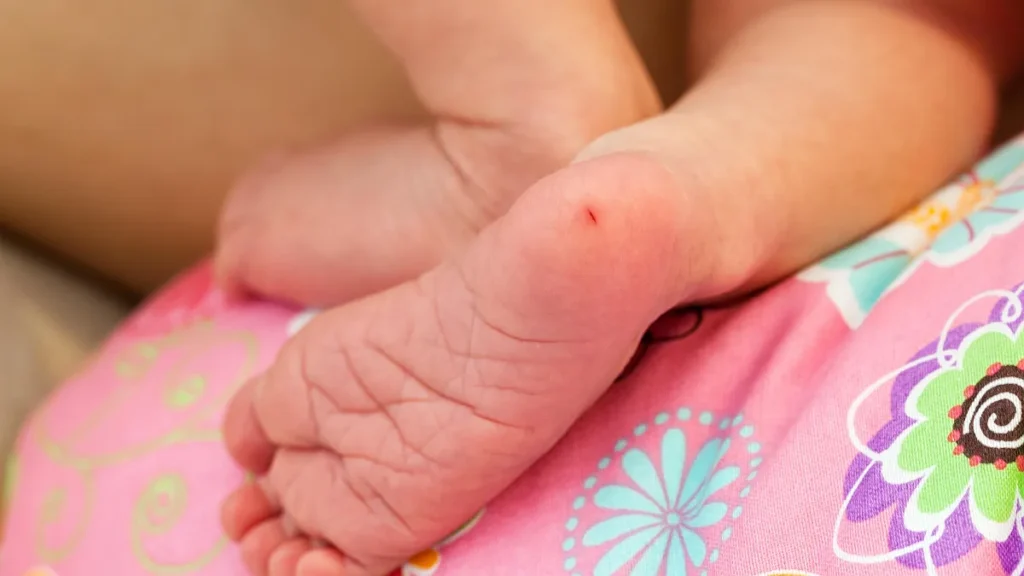
El diagnóstico se realiza principalmente mediante la prueba del tamiz neonatal, que consiste en obtener unas gotas de sangre del talón del recién nacido durante los primeros días de vida. Esta prueba permite identificar niveles elevados de fenilalanina antes de que aparezcan síntomas, facilitando un tratamiento temprano que previene las complicaciones más graves.
El tratamiento principal consiste en seguir una dieta estricta baja en fenilalanina durante toda la vida. Esto implica limitar o evitar alimentos ricos en proteínas y utilizar fórmulas especiales diseñadas para proporcionar los nutrientes necesarios sin exceso de fenilalanina. La dieta debe ser supervisada cuidadosamente por médicos y nutriólogos especializados.
Algunas personas con determinadas variantes de la enfermedad pueden beneficiarse de medicamentos que ayudan a mejorar la actividad de la enzima deficiente o a reducir los niveles de fenilalanina en la sangre. Sin embargo, la alimentación controlada sigue siendo la base fundamental del tratamiento.
El seguimiento médico regular es esencial para vigilar los niveles sanguíneos de fenilalanina y ajustar la dieta según la edad, el crecimiento y las necesidades individuales del paciente. Mantener niveles adecuados ayuda a preservar la función cerebral y a mejorar la calidad de vida.
Las mujeres con fenilcetonuria deben prestar especial atención durante el embarazo. Si los niveles de fenilalanina son elevados, pueden afectar gravemente al feto, incluso si el bebé no hereda la enfermedad. Por ello, es indispensable un control metabólico estricto antes y durante la gestación.
Gracias al diagnóstico temprano y al tratamiento adecuado, la mayoría de las personas con fenilcetonuria pueden llevar una vida normal, estudiar, trabajar y desarrollar sus actividades cotidianas sin limitaciones importantes. La detección oportuna mediante el tamiz neonatal ha transformado el pronóstico de esta enfermedad, evitando las graves consecuencias neurológicas que eran comunes antes de la implementación de estos programas de salud.
En conclusión, la fenilcetonuria es un trastorno metabólico hereditario que impide el procesamiento normal de la fenilalanina. Aunque puede causar daños severos al sistema nervioso si no se trata, el diagnóstico temprano y una dieta adecuada permiten que las personas afectadas tengan un desarrollo saludable y una buena calidad de vida.