Púrpura Trombocitopénica Idiopática (PTI)
La púrpura trombocitopénica idiopática (PTI), también conocida como trombocitopenia inmune primaria, es un trastorno hematológico caracterizado por una disminución anormal del número de plaquetas en la sangre. Las plaquetas son células fundamentales en el proceso de coagulación, y su déficit aumenta el riesgo de hemorragias.
Se denomina “idiopática” porque en muchos casos no se identifica una causa clara, aunque hoy se sabe que es un trastorno autoinmune: el propio sistema inmunológico del paciente produce anticuerpos que destruyen las plaquetas o interfieren con su producción en la médula ósea.
Causas y factores de riesgo
En la mayoría de los casos, no se encuentra un desencadenante evidente, pero existen situaciones que pueden preceder a la aparición del cuadro:
- Infecciones virales (hepatitis, VIH, varicela, citomegalovirus, Epstein-Barr).
- Vacunación reciente en algunos casos raros.
- Enfermedades autoinmunes asociadas.
- Más común en mujeres jóvenes y en la infancia después de infecciones virales.
Manifestaciones clínicas
Los síntomas dependen del grado de trombocitopenia:
- Equimosis y petequias: manchas rojas o moradas en la piel por pequeños sangrados.
- Epistaxis: sangrado nasal frecuente.
- Sangrado gingival: sobre todo al cepillarse los dientes.
- Menstruaciones abundantes (menorragia) en mujeres.
- En casos graves: hematuria (sangre en la orina), hemorragia digestiva o, raramente, hemorragia intracraneal.
Algunos pacientes pueden estar asintomáticos y la trombocitopenia descubrirse en un análisis de sangre rutinario.
Diagnóstico
No existe una prueba única que confirme la PTI, por lo que el diagnóstico es de exclusión. Se basa en:
- Biometría hemática: revela bajo número de plaquetas, con valores normales en glóbulos rojos y leucocitos.
- Frotis de sangre periférica: muestra plaquetas de tamaño normal o aumentado.
- Pruebas para descartar otras causas: infecciones virales, enfermedades autoinmunes, alteraciones de médula ósea.
Evolución
- En niños, suele ser aguda y autolimitada, resolviéndose en semanas o meses tras una infección viral.
- En adultos, tiende a ser crónica, con riesgo de recaídas y necesidad de tratamiento prolongado.
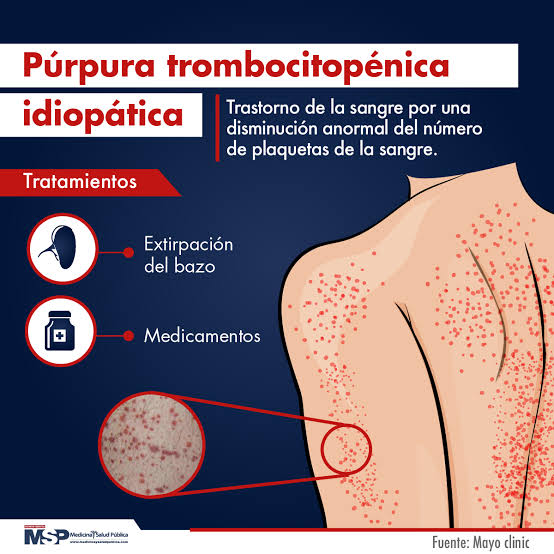
Tratamiento
El objetivo no es normalizar el número de plaquetas, sino prevenir hemorragias graves. La decisión depende de la severidad:
- Observación: si el paciente tiene más de 30,000 plaquetas y no hay sangrado significativo.
- Corticosteroides: como prednisona, primera línea de tratamiento para aumentar rápidamente el recuento plaquetario.
- Inmunoglobulina intravenosa (IVIG): útil en casos de sangrado grave o cuando se requiere un aumento rápido de plaquetas.
- Agonistas del receptor de trombopoyetina (eltrombopag, romiplostim): estimulan la producción de plaquetas en la médula ósea.
- Rituximab: terapia inmunosupresora en casos resistentes.
- Esplenectomía: opción en casos crónicos graves y refractarios, ya que el bazo es el principal sitio de destrucción de plaquetas.
Pronóstico
- En niños, el pronóstico es excelente, con resolución espontánea en la mayoría de los casos.
- En adultos, la enfermedad puede persistir y requerir terapias de mantenimiento, pero muchos pacientes logran un buen control.
- El riesgo principal son las hemorragias severas, que son poco frecuentes pero potencialmente mortales.