Miocardiopatía hipertrófica: causas, síntomas, diagnóstico, tratamiento y complicaciones
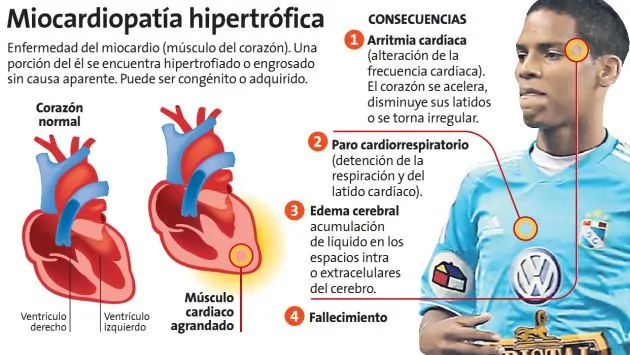
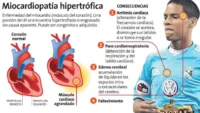
La miocardiopatía hipertrófica (MCH) es una enfermedad del músculo cardíaco caracterizada por un engrosamiento anormal de las paredes del corazón, especialmente del ventrículo izquierdo, que es la cavidad encargada de bombear la sangre hacia el resto del organismo. Este aumento del grosor no se debe a enfermedades como la hipertensión arterial o problemas en las válvulas cardíacas, sino que generalmente tiene un origen genético. Es una de las enfermedades cardíacas hereditarias más frecuentes y puede presentarse a cualquier edad, desde la infancia hasta la vejez.
En muchas personas la enfermedad puede permanecer sin síntomas durante años, mientras que en otras puede ocasionar molestias importantes e incluso aumentar el riesgo de arritmias graves y muerte súbita cardíaca, especialmente en personas jóvenes y deportistas. Sin embargo, gracias a los avances en el diagnóstico y tratamiento, la mayoría de los pacientes puede llevar una vida larga y con buena calidad si recibe seguimiento médico adecuado.
¿Qué ocurre en el corazón?
En la miocardiopatía hipertrófica, las fibras musculares del corazón crecen de manera desorganizada y el músculo cardíaco se vuelve más grueso de lo normal. Este engrosamiento puede dificultar el llenado del corazón durante la relajación (diástole) y, en algunos casos, obstruir parcialmente la salida de la sangre desde el ventrículo izquierdo hacia la aorta.
Además, las alteraciones estructurales favorecen la aparición de cicatrices microscópicas en el tejido cardíaco, lo que incrementa el riesgo de desarrollar arritmias.
Causas
La mayoría de los casos tiene un origen hereditario. Se debe a mutaciones en genes responsables de producir proteínas que forman el músculo cardíaco. Estas mutaciones suelen transmitirse de padres a hijos con un patrón autosómico dominante, lo que significa que cada hijo de una persona afectada tiene aproximadamente un 50 % de probabilidad de heredar la alteración genética.
En un pequeño porcentaje de casos, la enfermedad puede aparecer por una mutación espontánea sin antecedentes familiares conocidos.
También existen enfermedades que pueden producir un engrosamiento del corazón similar al de la miocardiopatía hipertrófica, como algunas enfermedades metabólicas o trastornos de almacenamiento, por lo que es importante diferenciarlas mediante estudios especializados.
Factores de riesgo
El principal factor de riesgo es tener antecedentes familiares de miocardiopatía hipertrófica, muerte súbita de origen cardíaco o arritmias hereditarias.
Otros factores que aumentan la probabilidad de detectar la enfermedad incluyen:
- Tener familiares con insuficiencia cardíaca de origen desconocido.
- Presentar desmayos inexplicables.
- Ser deportista competitivo con hallazgos anormales en estudios cardiológicos.
- Haber sido diagnosticado previamente con soplos cardíacos sin causa clara.
Síntomas
Muchas personas nunca presentan síntomas y descubren la enfermedad durante una revisión médica o al realizarse un electrocardiograma o ecocardiograma.
Cuando aparecen manifestaciones clínicas, las más frecuentes son:
- Falta de aire al realizar esfuerzo físico.
- Dolor u opresión en el pecho.
- Fatiga o cansancio excesivo.
- Mareos.
- Desmayos, especialmente durante el ejercicio.
- Palpitaciones.
- Sensación de latidos rápidos o irregulares.
- Disminución de la capacidad para realizar actividad física.
Los síntomas pueden empeorar con el ejercicio intenso, la deshidratación o situaciones que disminuyan la presión arterial.
Tipos
La enfermedad puede clasificarse según la presencia o ausencia de obstrucción al flujo de salida del ventrículo izquierdo.
La miocardiopatía hipertrófica obstructiva ocurre cuando el músculo engrosado dificulta el paso de la sangre hacia la aorta, siendo la forma más frecuente y la que suele provocar síntomas más intensos.
La miocardiopatía hipertrófica no obstructiva presenta engrosamiento del músculo cardíaco sin impedir significativamente la salida de la sangre.
Diagnóstico
El diagnóstico requiere una combinación de evaluación clínica, antecedentes familiares y estudios de imagen.
El médico realiza una exploración física donde puede detectar un soplo cardíaco característico.
Entre las pruebas más utilizadas se encuentran:
- Electrocardiograma.
- Ecocardiograma, considerado el estudio principal para confirmar el diagnóstico.
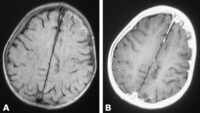
- Resonancia magnética cardíaca.
- Monitoreo Holter de 24 o 48 horas para detectar arritmias.
- Prueba de esfuerzo.
- Estudios genéticos cuando existe sospecha de enfermedad hereditaria.
- Estudios de familiares de primer grado mediante ecocardiograma y electrocardiograma.
Tratamiento
El tratamiento depende de la gravedad de los síntomas, el grado de obstrucción y el riesgo de presentar complicaciones.
Los medicamentos utilizados incluyen betabloqueadores y bloqueadores de los canales de calcio, que ayudan a disminuir la frecuencia cardíaca y mejorar el llenado del corazón. En algunos pacientes también pueden emplearse antiarrítmicos para controlar alteraciones del ritmo cardíaco.
En personas con obstrucción importante y síntomas persistentes, puede considerarse la miectomía septal, una cirugía en la que se retira parte del músculo engrosado para mejorar el flujo sanguíneo. Otra opción es la ablación septal con alcohol, procedimiento menos invasivo que reduce el grosor del músculo mediante la inyección controlada de alcohol en una arteria específica.
Los pacientes con alto riesgo de muerte súbita pueden requerir la implantación de un desfibrilador automático implantable (DAI), un dispositivo que detecta arritmias potencialmente mortales y administra una descarga eléctrica para restablecer el ritmo normal del corazón.
En casos avanzados de insuficiencia cardíaca que no responden al tratamiento, puede ser necesario un trasplante cardíaco.
Cambios en el estilo de vida
Las personas con miocardiopatía hipertrófica deben mantener un seguimiento periódico con el cardiólogo y cumplir estrictamente el tratamiento indicado.
También es recomendable evitar la deshidratación, controlar adecuadamente otras enfermedades como hipertensión o diabetes, mantener un peso saludable y consultar al especialista antes de practicar deportes competitivos o actividades físicas de alta intensidad.
En muchos casos se recomienda realizar ejercicio moderado supervisado, ya que la actividad física adaptada suele ser beneficiosa para la salud cardiovascular.
Complicaciones
Si no se controla adecuadamente, la enfermedad puede ocasionar complicaciones como:
- Insuficiencia cardíaca.
- Fibrilación auricular.
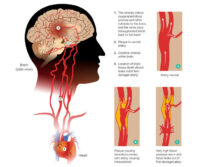
- Accidente cerebrovascular debido a la formación de coágulos.
- Arritmias ventriculares graves.
- Muerte súbita cardíaca.
- Endocarditis infecciosa en situaciones específicas.
- Dilatación progresiva del corazón en etapas avanzadas.
Pronóstico
El pronóstico ha mejorado considerablemente gracias al diagnóstico temprano y a los tratamientos actuales. La mayoría de los pacientes puede tener una esperanza de vida cercana a la de la población general si recibe seguimiento médico y se identifican oportunamente los factores de riesgo.
No obstante, algunas personas presentan un mayor riesgo de complicaciones, especialmente quienes tienen antecedentes familiares de muerte súbita, desmayos repetidos, arritmias ventriculares, un engrosamiento muy importante del músculo cardíaco o cicatrices extensas detectadas mediante resonancia magnética.
La detección temprana, el estudio de los familiares y el seguimiento por un cardiólogo son fundamentales para reducir el riesgo de complicaciones y mejorar la calidad de vida de quienes viven con esta enfermedad.