El síndrome de Hurler: una rara enfermedad hereditaria que afecta múltiples órganos
La mucopolisacaridosis tipo I (MPS I) es un trastorno hereditario raro, causado por la deficiencia de una enzima llamada alfa-L-iduronidasa, responsable de descomponer ciertas moléculas complejas conocidas como glucosaminoglucanos (GAGs). Esta deficiencia provoca la acumulación de GAGs en diversas células del cuerpo, lo que genera daño progresivo en múltiples órganos y tejidos. Cuando se presenta en su forma más severa, se conoce como síndrome de Hurler.
El síndrome de Hurler es parte de un espectro clínico que también incluye formas más leves como la MPS I tipo Scheie y la forma intermedia conocida como Hurler-Scheie. La forma Hurler es la más grave de todas, con síntomas que suelen aparecer durante el primer año de vida y progresar rápidamente si no se trata.
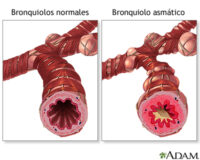
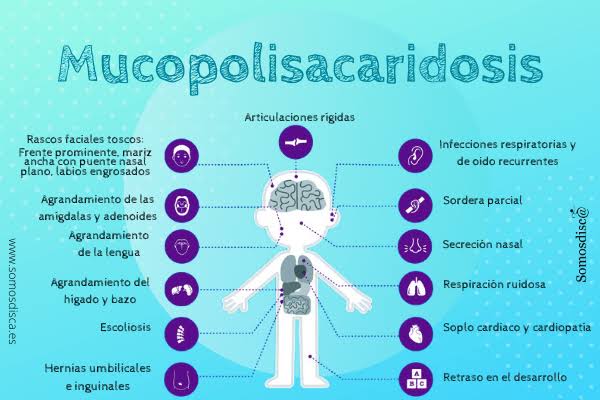
Los signos iniciales pueden ser sutiles, pero con el tiempo se desarrollan características físicas características: rasgos faciales toscos, cabeza grande (macrocefalia), puente nasal plano, lengua agrandada (macroglosia), y labios gruesos. Los bebés también presentan hernias umbilicales o inguinales, dificultad para alimentarse, infecciones respiratorias frecuentes, y retraso en el desarrollo motor.
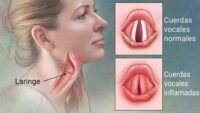
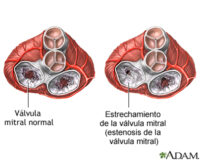
Conforme avanza la enfermedad, los niños desarrollan rigidez articular, baja estatura, deformidades esqueléticas (disostosis múltiple), agrandamiento del hígado y del bazo (hepatosplenomegalia), y alteraciones cardíacas. La córnea puede volverse opaca, lo que afecta la visión, y también pueden presentarse problemas auditivos debido a infecciones crónicas o daño a los huesecillos del oído.
Una de las manifestaciones más preocupantes es el retraso cognitivo, que suele hacerse evidente durante el segundo año de vida. Este deterioro progresivo lleva eventualmente a una discapacidad intelectual severa. Sin tratamiento, la expectativa de vida de quienes padecen el síndrome de Hurler es muy corta, usualmente de 10 años o menos, debido a complicaciones cardíacas o respiratorias.
El diagnóstico se basa en la evaluación clínica, análisis de orina (donde se detectan niveles elevados de GAGs), estudios enzimáticos que miden la actividad de la alfa-L-iduronidasa, y pruebas genéticas que confirman las mutaciones en el gen IDUA, responsable de codificar dicha enzima.
El tratamiento ha evolucionado en los últimos años. Uno de los pilares terapéuticos es la terapia de reemplazo enzimático (Terapia ERT), que consiste en la administración intravenosa de una forma sintética de la enzima deficiente. Este tratamiento puede mejorar varios síntomas físicos y la calidad de vida, aunque no cruza la barrera hematoencefálica, por lo que no detiene el deterioro neurológico.
Para abordar el compromiso del sistema nervioso central, el trasplante de células madre hematopoyéticas (generalmente de médula ósea o sangre del cordón umbilical) es la mejor opción si se realiza tempranamente, idealmente antes de los 2 años. Este procedimiento puede ayudar a preservar el desarrollo intelectual y mejorar la supervivencia, aunque conlleva riesgos importantes.
El manejo también incluye tratamientos de apoyo para las complicaciones respiratorias, ortopédicas, cardíacas y visuales, así como terapia ocupacional, fisioterapia y atención especializada en neurología, genética y pediatría.
Aunque el síndrome de Hurler no tiene cura definitiva, los avances médicos han permitido mejorar la calidad y la esperanza de vida de muchos pacientes. La detección temprana es clave, y por ello, algunos países ya incluyen la MPS I en sus programas de tamizaje neonatal.
El pronóstico depende del momento del diagnóstico, la severidad de los síntomas, y el acceso a tratamientos especializados. Con un enfoque multidisciplinario, es posible brindar a los pacientes y sus familias una mejor calidad de vida.