Distrofia miotónica: una enfermedad multisistémica hereditaria
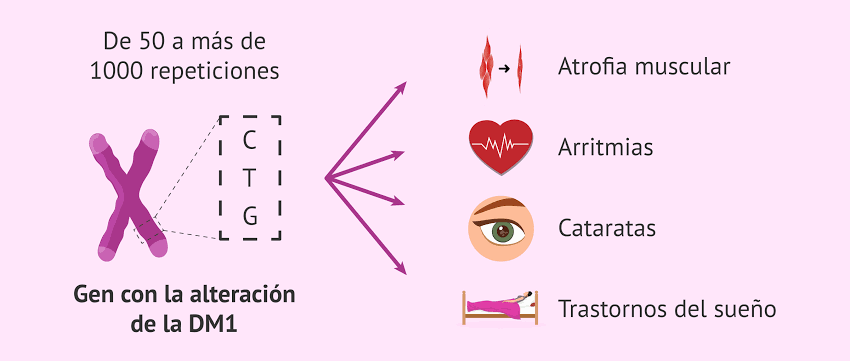
La distrofia miotónica es una enfermedad genética progresiva que afecta principalmente a los músculos, pero también puede comprometer diversos órganos y sistemas del cuerpo. Es la forma más común de distrofia muscular en adultos y se transmite con un patrón autosómico dominante, lo que significa que basta una copia del gen alterado para heredar la enfermedad.
Existen dos tipos principales:
- Distrofia miotónica tipo 1 (DM1 o enfermedad de Steinert): causada por una expansión del triplete CTG en el gen DMPK del cromosoma 19.
- Distrofia miotónica tipo 2 (DM2): menos grave y causada por una expansión CCTG en el gen CNBP (ZNF9).
Ambos tipos comparten características clínicas similares, aunque la DM1 tiende a ser más severa, con síntomas que pueden comenzar en la infancia, adolescencia o edad adulta, dependiendo de la magnitud de la mutación.
Una característica distintiva de la enfermedad es la miotonia, que consiste en la dificultad para relajar los músculos después de una contracción. Esto puede observarse, por ejemplo, cuando la persona tarda en soltar objetos después de agarrarlos.
Los síntomas musculares más frecuentes incluyen:
- Debilidad muscular progresiva, especialmente en extremidades distales (manos, pies)
- Dificultad para caminar o subir escaleras
- Caídas frecuentes
- Rigidez muscular (miotonia)
Además de los músculos, la enfermedad puede afectar numerosos órganos, por lo que se considera una enfermedad multisistémica. Entre las complicaciones más comunes se encuentran:
- Oculares: cataratas prematuras, a menudo antes de los 50 años
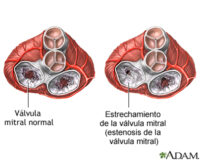
- Cardíacas: arritmias, bloqueos de conducción, riesgo de muerte súbita
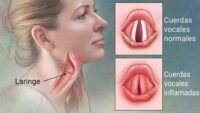
- Respiratorias: debilidad del diafragma, apnea del sueño, infecciones recurrentes
- Endocrinas: resistencia a la insulina, hipotiroidismo, alteraciones menstruales, infertilidad en hombres
- Sistema nervioso central: somnolencia excesiva, falta de concentración, apatía
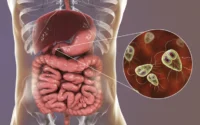
- Sistema digestivo: estreñimiento, dificultad para tragar (disfagia), alteración del vaciamiento gástrico
La forma congénita de la distrofia miotónica tipo 1 puede manifestarse desde el nacimiento con debilidad muscular severa, hipotonía, dificultades respiratorias y problemas de alimentación. Es la forma más grave y puede afectar el desarrollo motor e intelectual.
El diagnóstico se realiza mediante:
- Evaluación clínica y antecedentes familiares
- Electromiografía (EMG): para detectar la miotonía
- Pruebas genéticas: confirman la expansión anormal del triplete en el gen correspondiente
No existe cura para la distrofia miotónica, pero el tratamiento se enfoca en el manejo de los síntomas y en la prevención de complicaciones. Incluye:
- Medicamentos para la miotonía: como mexiletina
- Seguimiento cardiológico regular: con electrocardiogramas y posibles marcapasos
- Apoyo respiratorio: con dispositivos de presión positiva si hay apnea del sueño
- Fisioterapia y terapia ocupacional: para preservar la movilidad y la función
- Cirugías correctivas oculares en caso de cataratas
El pronóstico varía según la forma y la severidad. Algunos pacientes pueden tener síntomas leves y una vida casi normal, mientras que otros requieren cuidados constantes y presentan un deterioro significativo. Las formas congénitas tienen el peor pronóstico.
Es fundamental el asesoramiento genético, ya que cada hijo de una persona afectada tiene un 50% de probabilidad de heredar la mutación. También existen pruebas prenatales para detectar la enfermedad durante el embarazo.
La distrofia miotónica es una enfermedad compleja que requiere un abordaje multidisciplinario. A medida que avanza la investigación, se exploran tratamientos más específicos, como terapias dirigidas al ARN mutado y enfoques de edición genética, que podrían ofrecer opciones más efectivas en el futuro. Mientras tanto, el diagnóstico temprano y el seguimiento médico integral son claves para mejorar la calidad de vida de quienes la padecen.