Displasia arritmogénica del ventrículo derecho: el corazón que late con fragilidad
La displasia arritmogénica del ventrículo derecho (DAVD), también llamada miocardiopatía arritmogénica del ventrículo derecho (MAVD), es una enfermedad cardiaca hereditaria que altera la estructura y el funcionamiento del músculo cardíaco. Se caracteriza por la sustitución progresiva del tejido muscular del ventrículo derecho por grasa y tejido fibroso, lo que debilita las paredes del corazón y provoca arritmias potencialmente peligrosas. Aunque es poco frecuente, se trata de una de las principales causas de muerte súbita en personas jóvenes y deportistas.
Este trastorno fue descrito por primera vez en la década de 1980 y desde entonces ha sido objeto de un gran interés médico por su relación con las arritmias ventriculares malignas, que pueden desencadenar síncope o muerte súbita. Se estima que afecta entre 1 de cada 2,000 a 5,000 personas, aunque su prevalencia podría ser mayor debido a casos subdiagnosticados.
La DAVD tiene un origen genético y se hereda de manera autosómica dominante, lo que significa que basta con que uno de los padres sea portador de la mutación para que los hijos tengan riesgo de padecerla. Las mutaciones más comunes afectan genes relacionados con las proteínas desmosómicas, encargadas de mantener unidas las células del músculo cardíaco. Entre los genes más implicados se encuentran PKP2, DSG2, DSP, DSC2 y JUP. Estas mutaciones debilitan las uniones celulares y favorecen que el tejido muscular sea reemplazado por grasa y fibrosis.
El proceso de sustitución fibroadiposa del ventrículo derecho genera un terreno propicio para la aparición de arritmias ventriculares, que pueden ir desde extrasístoles hasta taquicardias o fibrilaciones ventriculares. Estas arritmias suelen desencadenarse durante el ejercicio físico intenso o situaciones de estrés, cuando el corazón se ve sometido a mayor demanda.
En cuanto a las manifestaciones clínicas, la enfermedad puede ser silenciosa durante años. Muchos pacientes permanecen asintomáticos hasta que presentan palpitaciones, mareos, pérdida de conciencia o incluso muerte súbita como primera manifestación. En otros casos, los síntomas iniciales incluyen fatiga, dolor torácico y disnea leve, especialmente durante el esfuerzo.
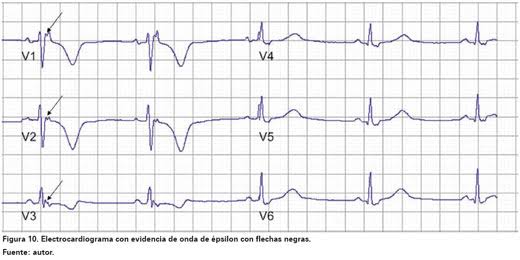
El electrocardiograma (ECG) suele mostrar alteraciones características, como ondas T invertidas en las derivaciones precordiales derechas o la presencia de un ondas épsilon, un pequeño potencial que aparece al final del complejo QRS y que refleja la conducción eléctrica anómala en el ventrículo derecho. Sin embargo, el diagnóstico requiere una evaluación integral, que puede incluir:
- Resonancia magnética cardíaca (RMC): permite visualizar la infiltración grasa y las áreas de fibrosis.
- Ecocardiograma: muestra la dilatación y el movimiento anormal del ventrículo derecho.
- Estudios genéticos: confirman la presencia de mutaciones en genes desmosómicos.
- Biopsia endomiocárdica: aunque se utiliza con menor frecuencia, puede demostrar el reemplazo fibroadiposo típico.
Los criterios diagnósticos internacionales (Task Force Criteria) combinan hallazgos estructurales, eléctricos, histológicos, familiares y genéticos para establecer el diagnóstico con mayor precisión.
El tratamiento de la displasia arritmogénica del ventrículo derecho se orienta a prevenir las arritmias malignas y evitar la muerte súbita. No existe una cura definitiva, pero el manejo médico y quirúrgico puede prolongar significativamente la vida del paciente. Entre las medidas terapéuticas más comunes se incluyen:
- Restricción del ejercicio físico intenso: especialmente el deporte competitivo, ya que aumenta el riesgo de arritmias.
- Medicamentos antiarrítmicos: como el sotalol o la amiodarona, para controlar las arritmias ventriculares.
- Desfibrilador automático implantable (DAI): en pacientes con alto riesgo de muerte súbita o antecedentes de arritmias graves.
- Ablación con catéter: en casos seleccionados para eliminar los focos de arritmia.
- Trasplante cardíaco: reservado para los casos más avanzados con insuficiencia cardíaca refractaria.
El pronóstico depende de la extensión del daño estructural y del control de las arritmias. Con un diagnóstico temprano y seguimiento adecuado, muchos pacientes pueden llevar una vida relativamente estable. Sin embargo, el riesgo de muerte súbita sigue siendo una preocupación importante, por lo que la vigilancia médica constante es esencial.
Desde el punto de vista preventivo, el estudio genético familiar es fundamental, ya que permite detectar portadores asintomáticos que pueden beneficiarse de controles periódicos o medidas preventivas antes de que se manifiesten los síntomas.
En el ámbito de la investigación, se están desarrollando terapias génicas y celulares para corregir los defectos moleculares responsables del daño en los desmosomas, aunque todavía se encuentran en etapas experimentales.
La displasia arritmogénica del ventrículo derecho es, en definitiva, una enfermedad en la que el corazón late con fragilidad, vulnerable al ritmo desordenado que impone la genética. Su estudio y comprensión han permitido salvar muchas vidas, pero continúa siendo un reto diagnóstico y terapéutico en la cardiología moderna. La clave para enfrentarlo sigue siendo la detección temprana, el control del esfuerzo físico y la vigilancia constante de quienes la padecen o tienen riesgo de desarrollarla.