Síndrome de Marfan
El síndrome de Marfan es un trastorno genético del tejido conectivo caracterizado por la afectación multisistémica de estructuras como el sistema cardiovascular, el esqueleto y los ojos. Se trata de una enfermedad de herencia autosómica dominante cuya gravedad varía ampliamente entre individuos, incluso dentro de la misma familia. Su principal riesgo deriva de las complicaciones cardiovasculares, especialmente la dilatación progresiva de la aorta y la posibilidad de disección aórtica.
Definición y bases genéticas
El síndrome de Marfan es causado en la mayoría de los casos por mutaciones en el gen FBN1, localizado en el cromosoma 15. Este gen codifica la fibrilina-1, una proteína esencial para la formación de microfibrillas, componentes fundamentales del tejido conectivo. La alteración de esta proteína genera una debilidad estructural en ligamentos, vasos sanguíneos y otros tejidos elásticos.
La herencia es autosómica dominante, lo que significa que una sola copia del gen mutado es suficiente para producir la enfermedad. Aproximadamente el 75% de los casos corresponden a individuos con un progenitor afectado, mientras que alrededor del 25% se originan por mutaciones de novo en personas sin antecedentes familiares.
Manifestaciones clínicas
El síndrome de Marfan presenta un espectro clínico amplio. Sus manifestaciones se agrupan principalmente en tres sistemas: cardiovascular, esquelético y ocular, aunque puede involucrar otros órganos.
Sistema cardiovascular
Es el sistema más relevante desde el punto de vista pronóstico. Las características más importantes son:
- Dilatación de la aorta ascendente, especialmente en la raíz aórtica.
- Aneurisma aórtico predispuesto a rotura.
- Disección aórtica, que constituye la complicación más grave y potencialmente mortal.
- Prolapso de la válvula mitral, a menudo acompañado de regurgitación mitral.
- En algunos casos, afectación de la válvula aórtica o insuficiencia aórtica.
Sistema musculoesquelético
Las manifestaciones esqueléticas son muy características y a menudo orientan al diagnóstico:
- Talla elevada desproporcionada con extremidades largas.
- Araquidactilia (dedos largos y delgados).
- Escoliosis o cifoescoliosis.
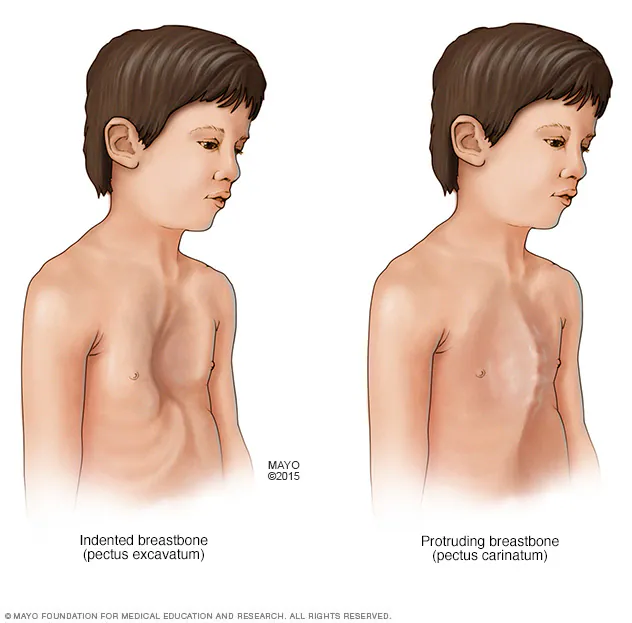
- Deformidades torácicas como pectus excavatum o pectus carinatum.
- Hiperlaxitud articular y tendencia a luxaciones.
- Paladar alto y estrecho, pie plano y otras alteraciones posturales.
Sistema ocular
Las alteraciones más frecuentes son:
- Ectopia lentis (subluxación o desplazamiento del cristalino), considerada un signo clave.
- Miopía elevada de inicio temprano.
- Riesgo aumentado de desprendimiento de retina.
- Cataratas o glaucoma en etapas avanzadas.
Manifestaciones adicionales
El síndrome también puede afectar otros órganos y tejidos:
- Sistema respiratorio: mayor riesgo de neumotórax espontáneo.
- Piel: presencia de estrías atróficas sin relación con cambios de peso.
- Sistema nervioso central: duralectasia (dilatación del saco dural), que puede producir dolor lumbar o neuropatías.
- Hernias y debilidad general del tejido conectivo.
Diagnóstico
El diagnóstico se basa en una combinación de criterios clínicos, familiares y genéticos. Actualmente se utilizan los criterios de Ghent revisados (2010), que consideran elementos cardinales como:
- Dilatación de la raíz aórtica.
- Ectopia lentis.
- Mutación patogénica confirmada en el gen FBN1.
- Conjunto de rasgos sistémicos valorados mediante una puntuación específica.
Para confirmar el diagnóstico se realizan:
- Ecocardiograma para evaluar la aorta y las válvulas.
- Examen oftalmológico con lámpara de hendidura.
- Radiografías o estudios ortopédicos para alteraciones esqueléticas.
- Pruebas genéticas para detectar mutaciones en FBN1.
Tratamiento y manejo
El síndrome de Marfan no tiene cura, pero su manejo adecuado reduce de manera significativa las complicaciones y mejora la supervivencia.
Tratamiento farmacológico
- Betabloqueantes como el propranolol o atenolol, utilizados para disminuir la tensión sobre la pared aórtica.
- Antagonistas de los receptores de angiotensina II (como losartán), que han demostrado beneficios en la reducción de la progresión de la dilatación aórtica.
Intervenciones quirúrgicas
Se consideran en casos de dilatación aórtica significativa o síntomas valvulares avanzados. Incluyen:
- Cirugía de reemplazo de la raíz aórtica o reparación preventiva antes de que la aorta alcance un tamaño crítico.
- Cirugías ortopédicas o torácicas según la gravedad de las deformidades.
- Intervenciones oftalmológicas en casos de ectopia lentis complicada o glaucoma.
Recomendaciones generales
- Evitar actividades físicas de alto impacto o levantamiento de peso excesivo.
- Seguimiento cardiológico periódico, generalmente anual o más frecuente según la evolución de la aorta.
- Revisión oftalmológica regular.
- Consejería genética para pacientes y familiares.
Pronóstico
Con los avances en el diagnóstico temprano, el tratamiento farmacológico y la cirugía preventiva, la esperanza de vida de las personas con síndrome de Marfan se ha incrementado significativamente. Con seguimiento médico adecuado y control riguroso de los factores de riesgo, los pacientes pueden llevar una vida relativamente normal y con buena calidad.