Osteogénesis imperfecta
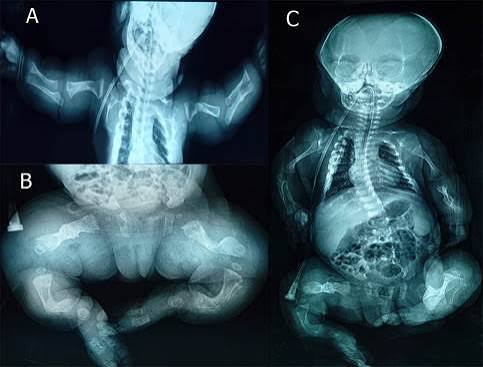
La osteogénesis imperfecta (OI) es un trastorno genético poco frecuente caracterizado principalmente por una fragilidad ósea anormal que provoca fracturas repetidas con mínimos traumatismos o incluso de manera espontánea. Popularmente se le conoce como la “enfermedad de los huesos de cristal”, debido a que los huesos son mucho más frágiles que lo normal.
Esta enfermedad está causada por mutaciones en los genes responsables de la producción del colágeno tipo I, una proteína esencial para la resistencia y elasticidad de los huesos y otros tejidos. Los genes más implicados son COL1A1 y COL1A2, cuya alteración puede llevar a una síntesis reducida de colágeno o a la producción de colágeno anormal. La herencia es generalmente autosómica dominante, aunque en algunos casos existen formas autosómicas recesivas y mutaciones de novo, lo que explica la variabilidad en su presentación clínica.
Los síntomas y la gravedad de la osteogénesis imperfecta son muy variables, desde formas leves con pocas fracturas a lo largo de la vida, hasta formas graves que se manifiestan desde el periodo neonatal con múltiples fracturas y deformidades esqueléticas. Entre las manifestaciones más características se encuentran:
- Fracturas frecuentes con traumatismos mínimos.
- Deformidades óseas progresivas, especialmente en extremidades largas y columna.
- Baja estatura, resultado de la afectación del crecimiento óseo.
- Escleróticas azules, un signo clásico debido a la delgadez de la esclerótica que deja ver el color subyacente de la coroides.
- Dentinogénesis imperfecta, que ocasiona dientes frágiles, decolorados y con tendencia a desgastarse o romperse.
- Hipoacusia progresiva, que aparece en la adolescencia o adultez temprana por alteración en los huesos del oído medio.
- Hiperlaxitud ligamentosa y debilidad muscular, que contribuyen a la inestabilidad articular.
Clínicamente, la OI se clasifica en diferentes tipos según la severidad, siguiendo la clasificación de Sillence, que reconoce desde la forma leve (tipo I) hasta formas letales en la infancia (tipo II). Los tipos III y IV presentan gravedad intermedia con deformidades importantes y fracturas múltiples a lo largo de la vida. En los últimos años, se han descrito nuevos tipos asociados a mutaciones en otros genes, lo que ha ampliado la clasificación.
El diagnóstico se basa en la historia clínica, el antecedente familiar y los hallazgos físicos. Se utilizan estudios de imagen, principalmente radiografías, que muestran fracturas en diferentes etapas de consolidación, huesos delgados y deformados. En algunos casos se realizan pruebas genéticas para confirmar la mutación y ofrecer asesoramiento genético a las familias.
No existe una cura definitiva para la osteogénesis imperfecta, por lo que el tratamiento se centra en mejorar la calidad de vida, prevenir complicaciones y favorecer la autonomía. El abordaje incluye varias estrategias:
- Tratamiento farmacológico: el uso de bisfosfonatos intravenosos (como pamidronato) ha mostrado eficacia en el aumento de la densidad mineral ósea y en la reducción de fracturas en pacientes pediátricos.
- Fisioterapia y rehabilitación: fundamentales para mejorar la fuerza muscular, la movilidad y la independencia funcional, evitando la atrofia por inmovilización prolongada.
- Cirugía ortopédica: en casos de fracturas recurrentes o deformidades severas se utilizan clavos intramedulares telescópicos que refuerzan los huesos largos y corrigen la alineación.
- Cuidados odontológicos: para el manejo de la dentinogénesis imperfecta y la preservación de la función masticatoria.
- Apoyo auditivo: uso de auxiliares auditivos o cirugía en caso de hipoacusia significativa.
- Asesoramiento genético: esencial para las familias, dado que se trata de una enfermedad hereditaria.
El pronóstico depende del tipo de osteogénesis imperfecta. Las formas leves permiten una expectativa de vida prácticamente normal, aunque con limitaciones físicas y necesidad de controles médicos frecuentes. Las formas moderadas pueden generar discapacidad significativa debido a las múltiples fracturas y deformidades, mientras que las formas más graves, como la tipo II, suelen ser incompatibles con la vida en los primeros meses.
Más allá de los aspectos médicos, la osteogénesis imperfecta también tiene un fuerte impacto emocional y social, tanto en los pacientes como en sus familias. La dependencia de cuidados constantes, las hospitalizaciones frecuentes y la necesidad de adaptar el entorno para prevenir caídas generan un desafío continuo. La educación del paciente y de su entorno es clave para promover la integración escolar, social y laboral, además de fomentar la independencia dentro de las posibilidades individuales.
En síntesis, la osteogénesis imperfecta es una enfermedad genética de gran variabilidad clínica que afecta el tejido óseo y otros órganos relacionados con el colágeno. Aunque no tiene cura, los avances en el tratamiento médico y quirúrgico han mejorado notablemente la calidad de vida de quienes la padecen, permitiendo que muchos pacientes lleven una vida activa con el acompañamiento adecuado de un equipo multidisciplinario.